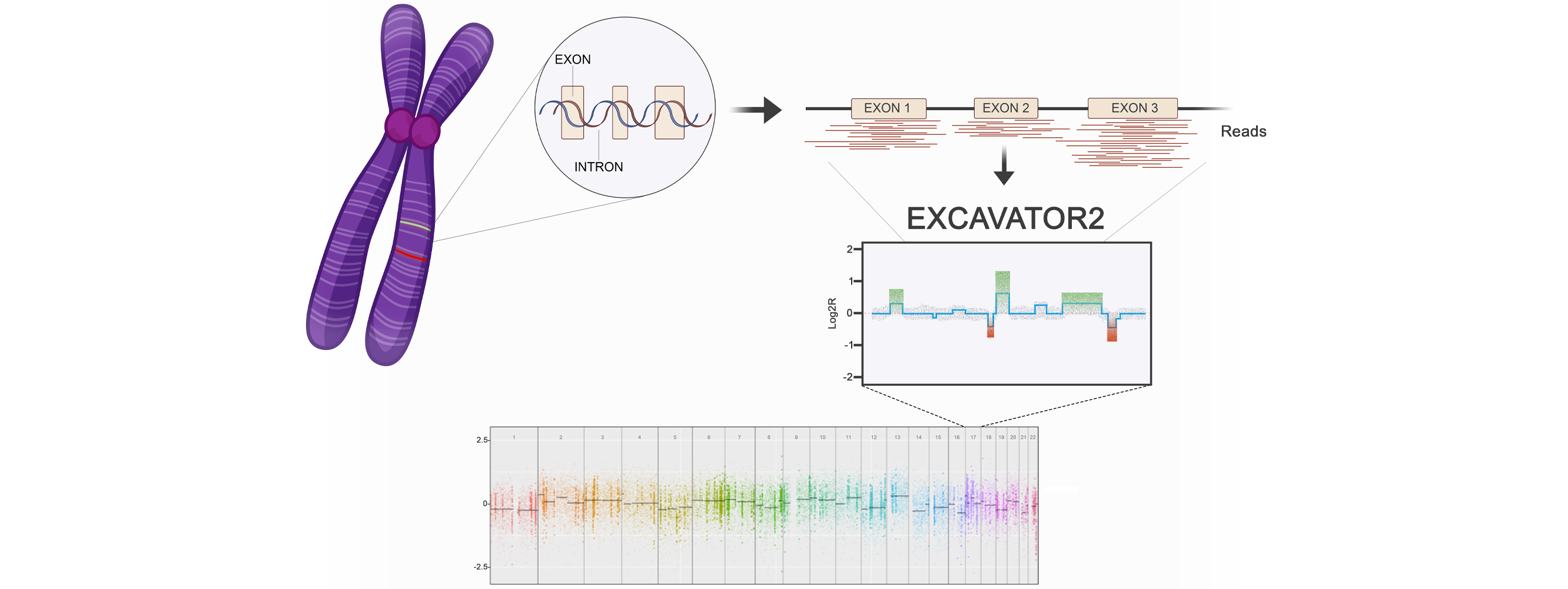
We develop computational methods for the identification of copy number variants (CNVs), which refer to the presence of an abnormal number of copies of a particular DNA segment. The fraction of the human genome affected by CNVs is substantial (5-10%) and contributes to phenotypic variation, but it also impacts several diseases, including cancer, cardiovascular disease, HIV acquisition and progression, autoimmune diseases, and Alzheimer’s and Parkinson’s diseases in complex and as yet unexplored ways. We are particularly dedicated to the optimisation of models for the clinical setting, both for diagnostic purposes and for a better characterisation of tumour heterogeneity, using ngs-based assays (whole exome and gene panel).