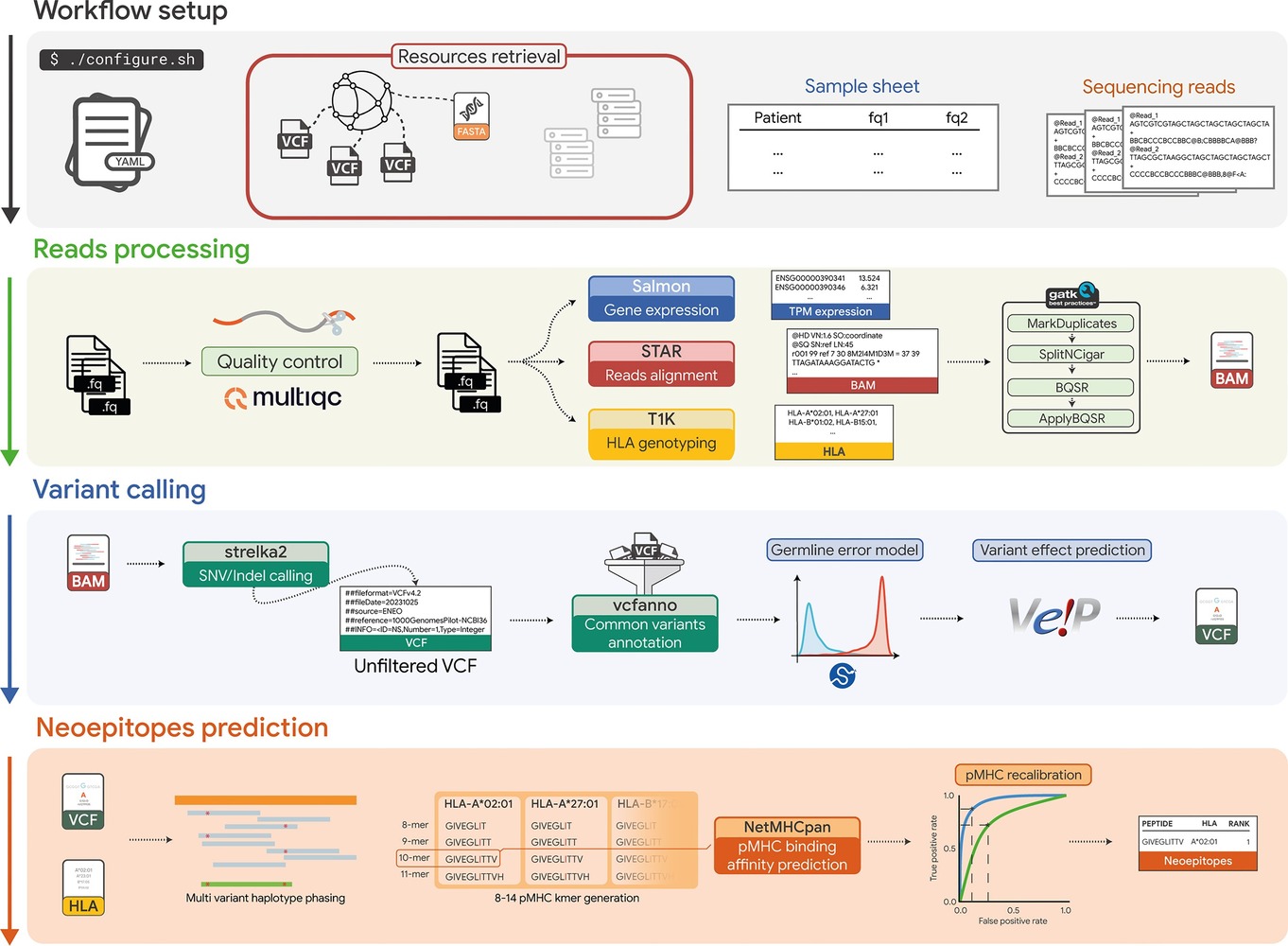
Neoepitopes arising from somatic mutations are capable of triggering host immune response which may result in tumor eradication. Their private nature makes it necessary to develop individualized detection and manufacturing processes, resulting in a long and expensive process. In this setting, avoiding false positives while keeping high sensitivity is key. To date multiple ongoing trials have focused on the screening of genomic data (WES/WGS) for the detection of somatic alterations, requiring at the meantime transcriptomic data (RNA-seq) to confirm the expression of the mutated transcript. Motivated by the growing evidence pointing to the impossibility of identifying every event responsible for the generation of MHC presented peptides, we seek to investigate the applicability of detecting immunogenic peptides in tumor samples using just the tumor RNAseq data, without relying on control data. We then implemented an end-to-end computational workflow which tunes state of art methods and uses a germline error model to distinguish germline variants from thus of somatic provenience. We assessed the performances across different tumor types using publicly avilable data, demonstrating its capability to correctly identify immunogenic peptides without requiring healthy matched controls.